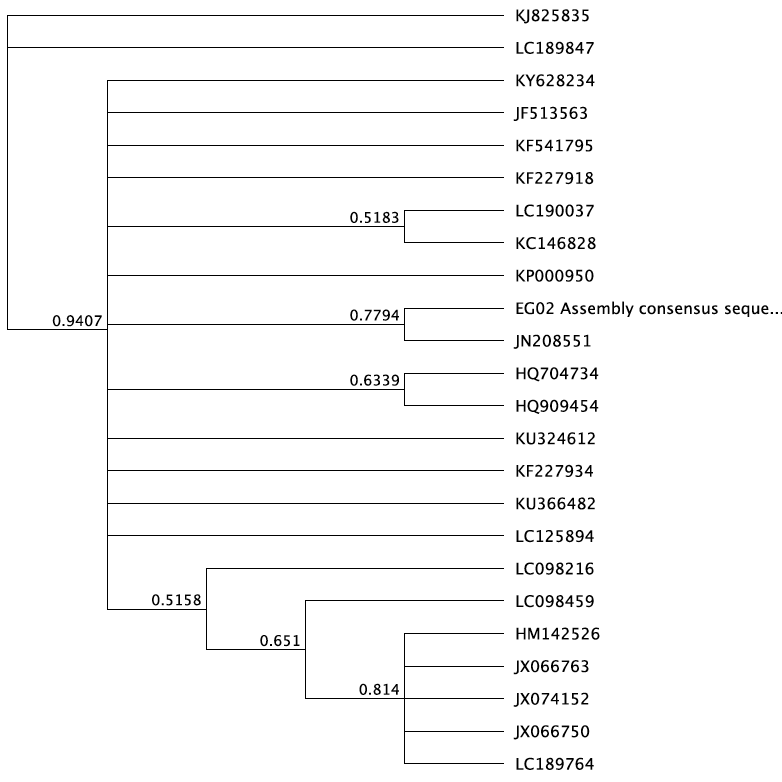
Using our fish DNA consensus sequences, along with around 25 other fish species sequences and an outgroup (I used a hammerhead shark), that has the CO1 gene, we created multiple phylogenies using different statistical methods. We cleaned up the DNA first, making sure that they all start and end at the same point. We tested out different methods: the Bayesian inference (GTR substitution model). We ran this twice, one for a short period and another one for a long period to see the differences that time could give us. We also used the Maximum Likelihood via rapid bootstrap and rapid hill climbing. The methods produced different consensus trees. For the final tree, we used the MrBayes method (HKJ model) for around 1,100,000 base pairs.