We scored our ISSR gels for two molecular markers: Omar and 17898, on an excel spreadsheet. If there is a band present, we marked it with an 1, if not it’s marked with a 0. I used the short run for Omar and the long run for 17989 because they looked like the bands were easier to see. Then we converted this into a Nexus file. The photos are attached at the bottom!



In lab, we did two Geneious tutorials. One was for a Map-to-reference, and the other was for a De-novo assembly.
Map to Reference tutorial
For the map-to-reference, we learned how to sequence map and SNP calling. We prepared the data by trimming their ends, assembled them, exploring the contigs, how to find SNPs and how to compare them. 5 reads had their ends trimmed due to low quality, they were 55687/1, 135904/1, 96204/1, 145868/1 and 76618/2. It took about 5-10s for the reads to map to the reference genome. There was least coverage at the ends with an average of 98% being covered and the max being 139. When I changed from the highest quality settings to 100% identical, from afar, it looked like there wasn’t any change but if you looked closely, there were a few sites that changed (e.g. site 28, 38, 41 and 73), which shows that there could’ve been a polymorphic site there. The end regions had >2 stdev below the means and we want to exclude them because I assume it’s not reliable? There were some transition sites (site 1554 and 1935), and transversions sites (site 1557 and 1923). I’m not sure if they affected the protein or not. 4 SNPs were excluded from the region. Below is my sequence view of my annotated reference genome with SNP calls along with my polymorphism table (well a chunk of it, it’s a huge file I can’t fit it all in).


De Novo Assembly
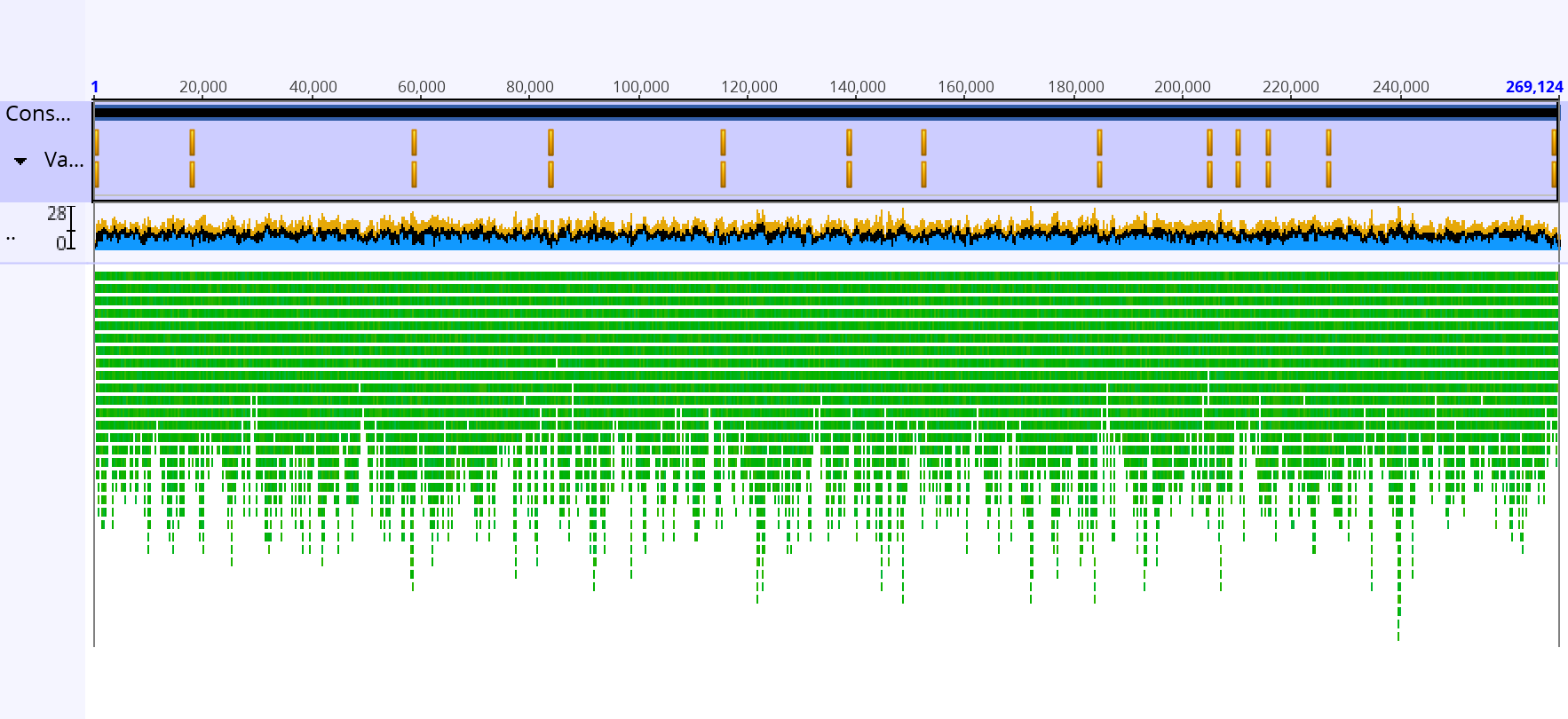
We assembled a bacterial gene in a 3 step tutorial. We assembled the short reads data then we assembled the reads using paired-end information and then we looked at the consensus sequence and tried to fix it. We assembled 2 reads and got back 4 contigs. The mean length of contigs >1000bp is about 130k bp, the shortest contig was around 512 bp. the NC50 score was 192, 891. The De Novo assembly, with and without paired ends, took roughly a minute to complete. There are some sites (15903, 284467) that had no coverage. The final length of the consensus sequence is 269,124bp. The consensus sequence is below.![]()