Carter Pope
Professor John Paul
Molecular Ecology
11/20/18
Lab 12 Entry
ISSR Analysis
On November 14th I arrived at the lab and began by adding 1 microliter of loading dye to each of my 15 PCR tubes with a micropipette. I then went over to a large 2 % gel and a lab mate and I (Jinwoo) transferred 5 microliters of each of our samples into individual wells, making sure to change out pipette tips each time and that we recorded where our specific samples were located on the large gel. Once everyone in the lab transferred their samples into the large gel, the gel was run for a long time on a low voltage.
I then turned on my laptop and loaded Geneious up onto my screen. Unfortunately, our lab table’s DNA showed no success, so our professor had us use a different set of DNA and assigned us with a new marker (5551) to use as a replacement. Since we got new DNA and marker, we redid steps from a previous lab dealing with coding heterozygotes. I made a new folder in Geneious, downloaded, and extracted the DNA set for ‘5551’ from canvas to my folder and made an alignment using ‘Muscle’. I edited any bad parts of the alignment and then located any heterozygotes that needed new ambiguity codes. The next thing I did was select each forward and reverse pair for a single sample and make a consensus sequence for each one. After making sure each of these had correct editings and base calls, I created a sequence list with all of the consensus sequences that I made with each pair. Then I added in the two samples that didn’t have a pair (JP1314 and TG0248) with the sequence list and made an alignment with all of this using ‘Muscle’. Making sure edits were made on any necessary trimmings of the alignment, I then named it “5551_Nucleotide alignment_Carter” and put this into a new folder labeled as “Concatenate.” (I must’ve read past the next step during lab, so a few days later I exported this alignment from Geneious into canvas under the assignment called “EPIC ALIGNMENTS”)
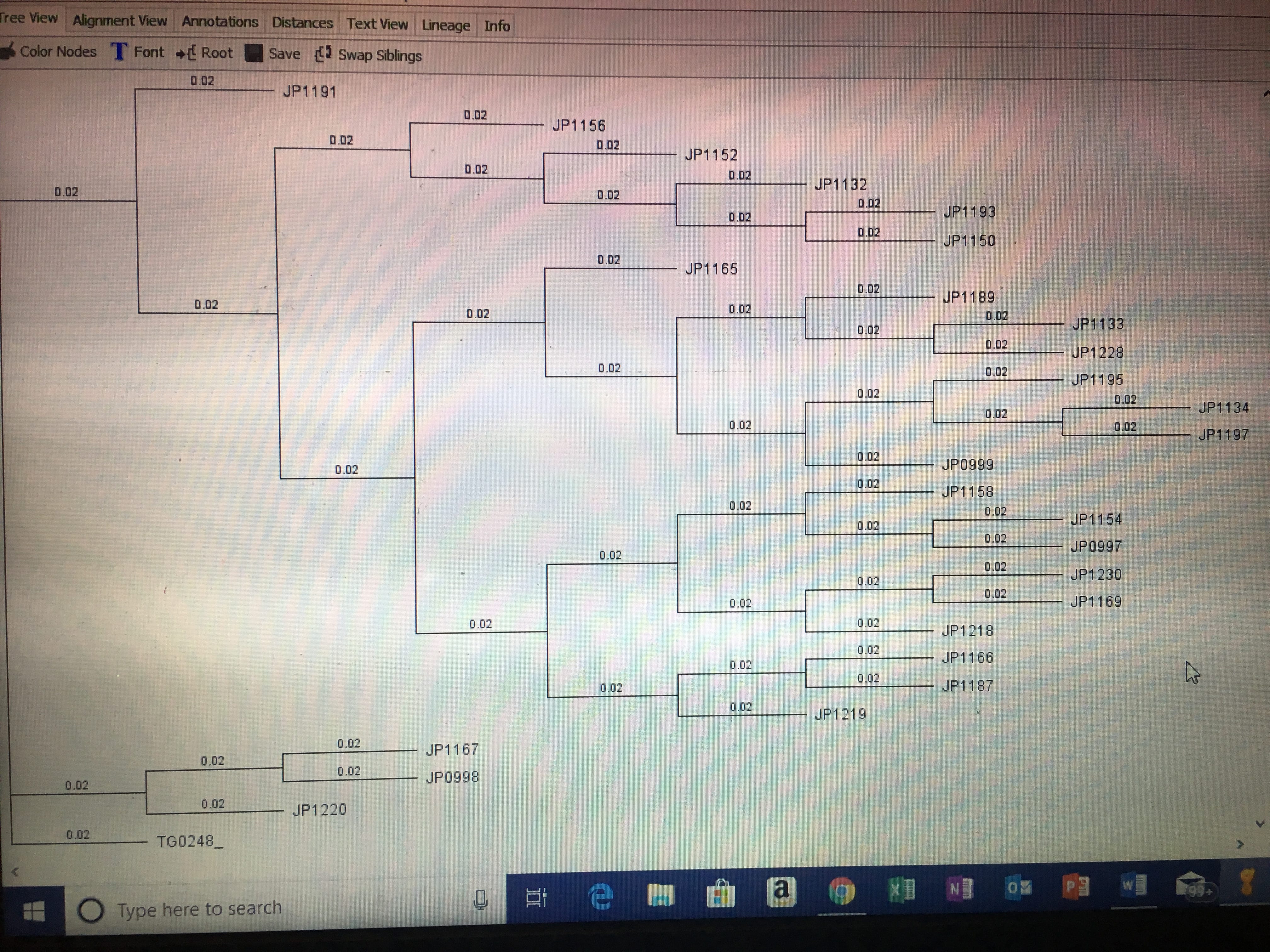
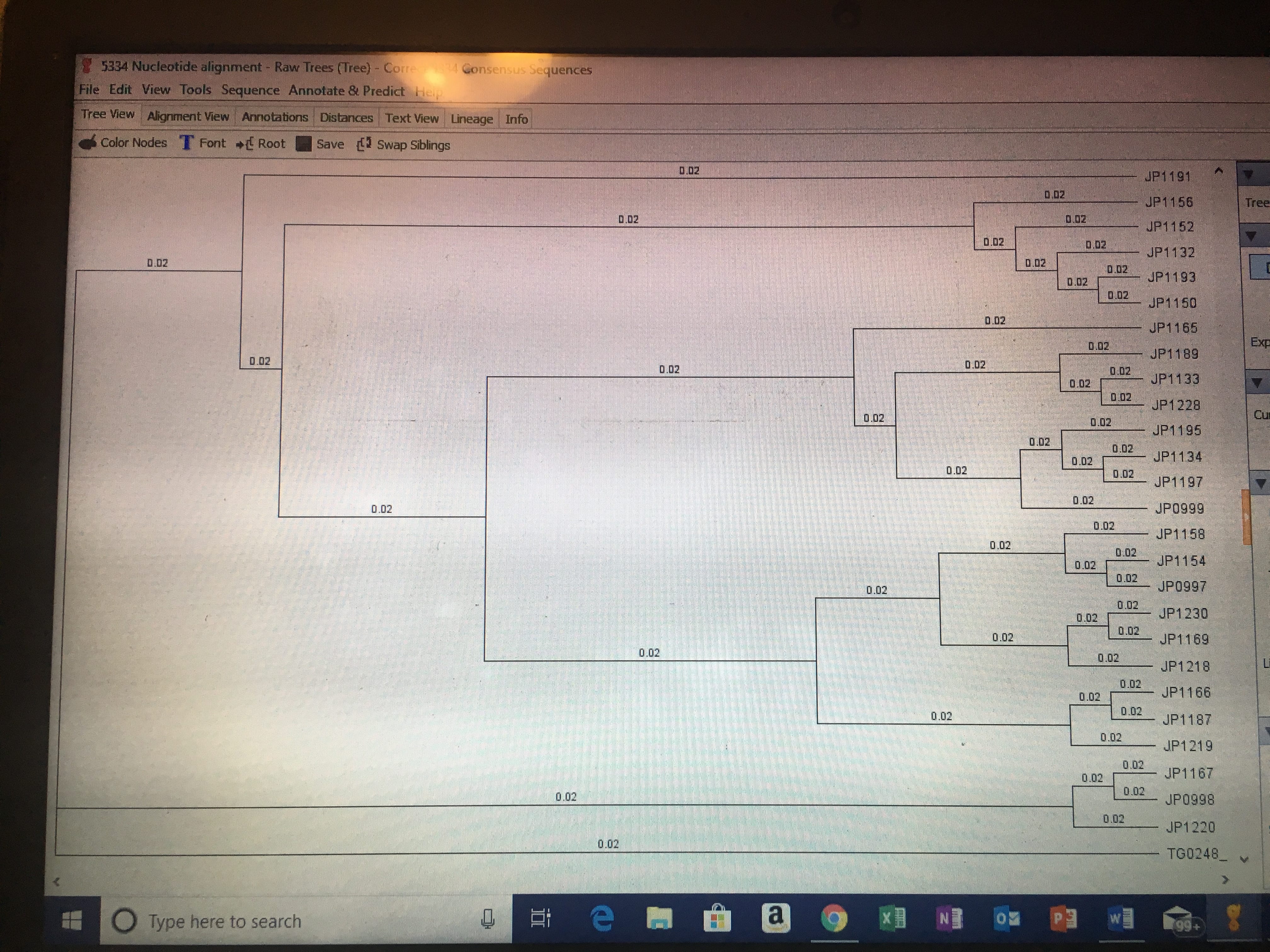
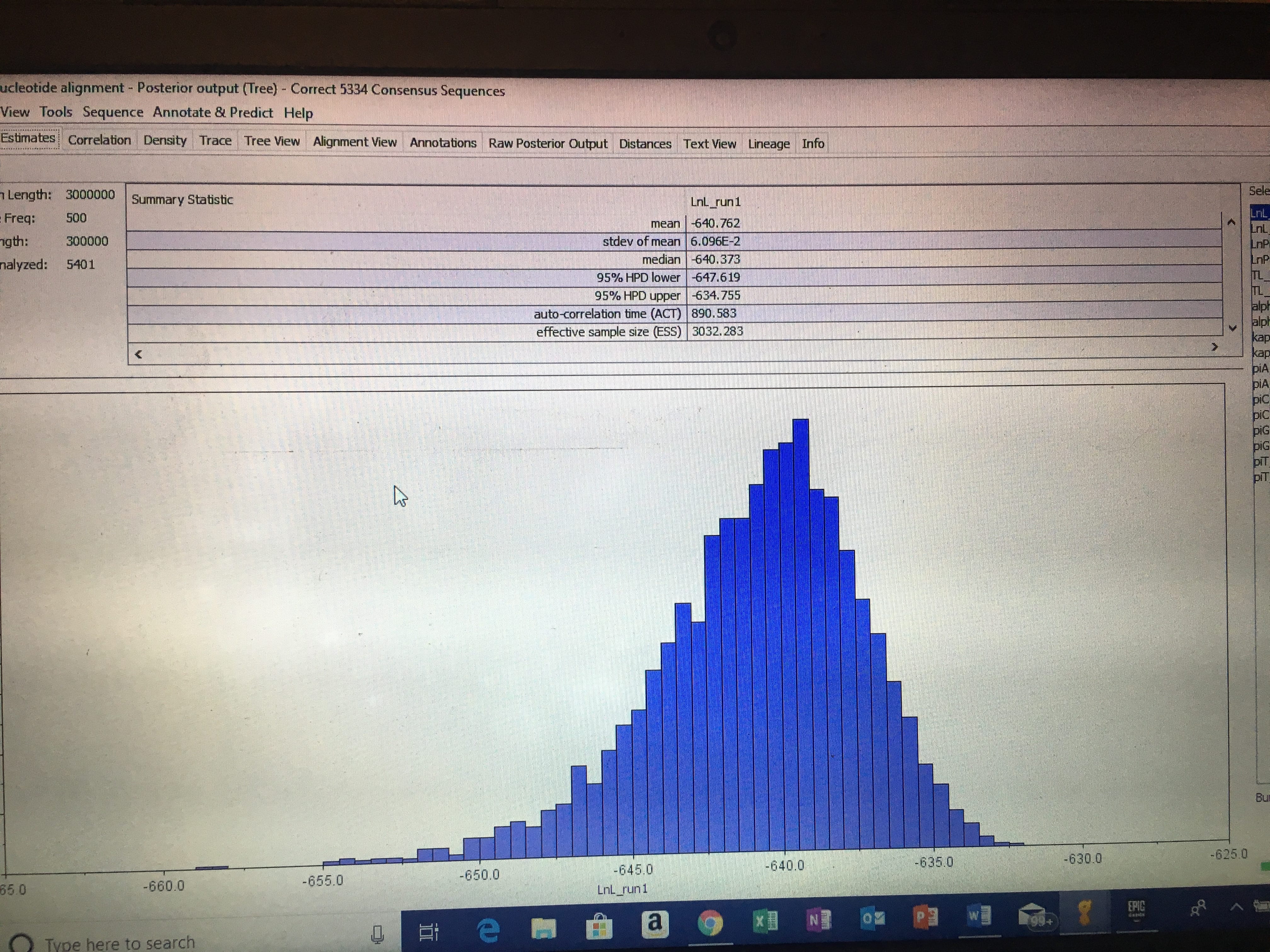
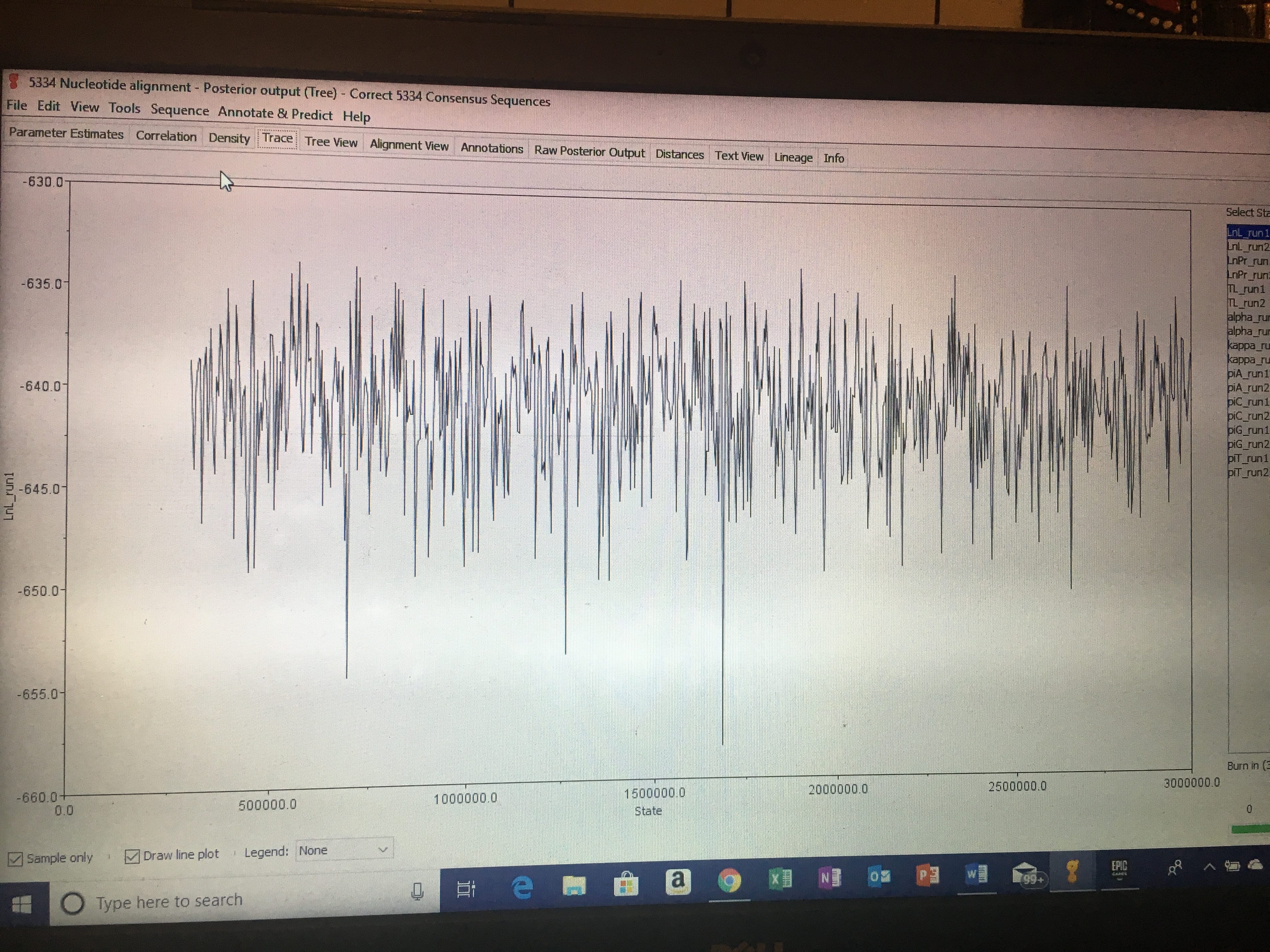
I then went to canvas and chose three other marker alignments (making neither of them were from the same marker) and downloaded/exported them into my Geneious folder called “Concatenate.” I then selected these three marker alignments, along with mine, and made a concatenate alignment. Further use of this concatenate alignment will be used to create analyses and phylogeny trees in further assignments.