I created an alignment of 25 COI sequences from Actinopterygii and my fish DNA barcode sequences, including one Chondrichthyes sequence as an out-group. I did this by searching COI and Actinopterygii/Chondrichthyes in the NCBI nucleotide program in Geneious. I chose sequences of similar lengths to my fish DNA sequences. I edited the alignment by choosing ‘Allow editing’ so that the sequences began and ended at the same point. When looking at the first 20 columns of my alignment, there were nine polymorphisms.
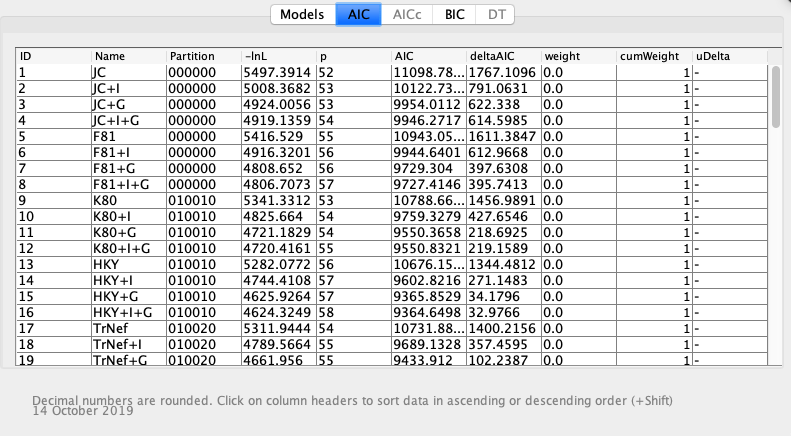
Best model based on AIC.

Best model based on BIC.
Then, I downloaded jModelTest2 to determine the best model of molecular evolution for my sequences. In Geneious, I exported the alignment in Philip format (relaxed). Then, in jModelTest2, I opened the file by clicking ‘File’ and ‘Load DNA Alignment’. Then I clicked ‘Analysis ‘and ‘Compute likelihood scores’, keeping the default settings. I looked at two methods, Akaike Information Criterion (AIC) and Bayesian Information Criterion (BIC). I did this by clicking ‘Do AIC/BIC’ respectively under the analysis window, keeping the default settings, and clicking ‘Do AIC/BIC calculations’. The best model based on AIC was JC, which was the same as the best model based on BIC.
Next, using Bayesian inference in Geneious, I selected my alignment, right clicked and chose ‘Tree’, then ‘MrBayes’. I used JC69 for the ‘substitution model’ and equal for ‘rate variation’. I kept the ‘gamma categories’ selection at 4. The ‘outgroup’ was put on HM422916, the sandshark outgroup I found earlier. For the initial ‘chain length’, I set it to 110,00. The ‘burn-in length’ was 10,000. I used the default setting for all other parameters. After running this analysis, I opened the ‘posterior output’ line and clicked on the ‘parameter estimates’ and ‘trace’ tabs.
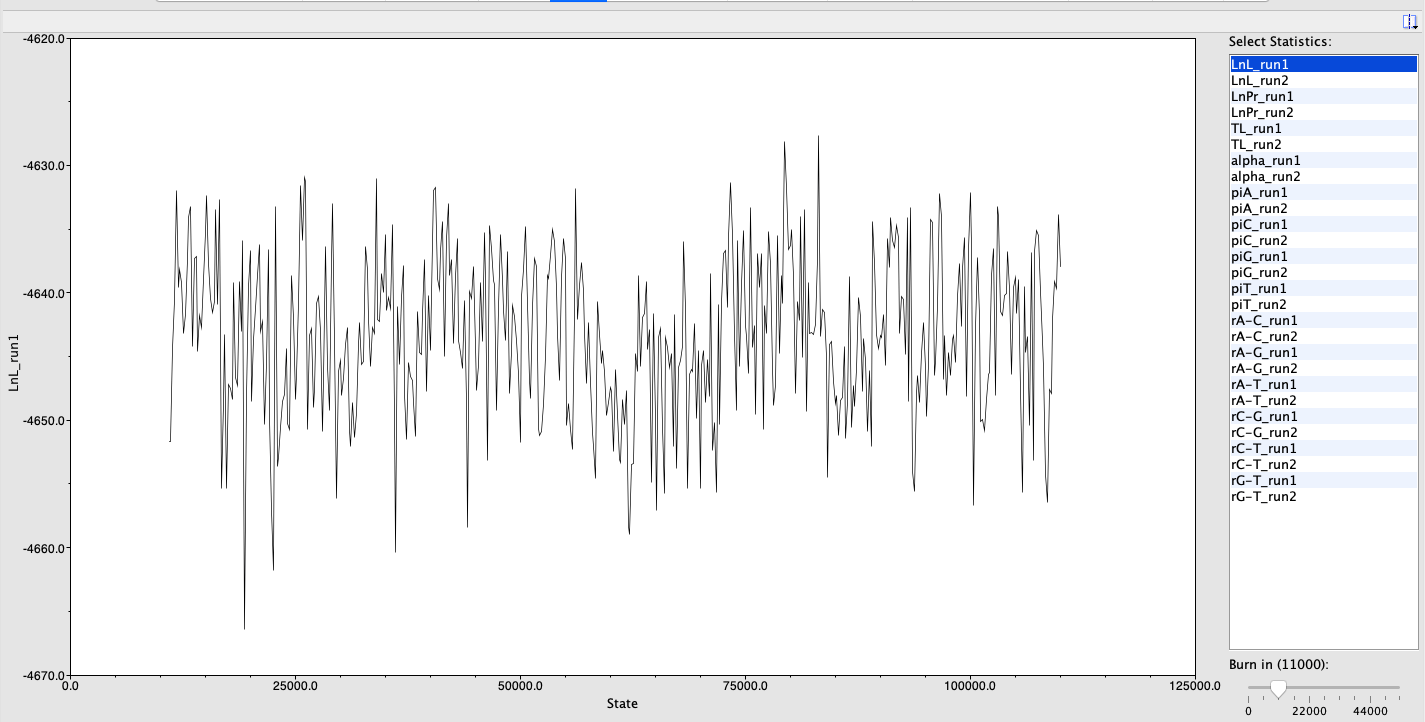
Trace of MrBayes chain length 110,000

Parameter estimates of MrBayes chain length 110,000.
Next, using maximum likelihood to infer a phylogenetic tree, I installed RAxML. I chose a similar evolutionary model to the MrBayes model and chose ‘rapid bootstrap with rapid hill climbing’. Once RAxML was done, I right-clicked the new line and chose ‘Tree’ and then ‘Consensus Tree Builder’. Then, I clicked ‘create consensus tree’ and ‘support threshold’ of 50%. The clades in the resulting tree did not match the ones from the Bayesian analysis.
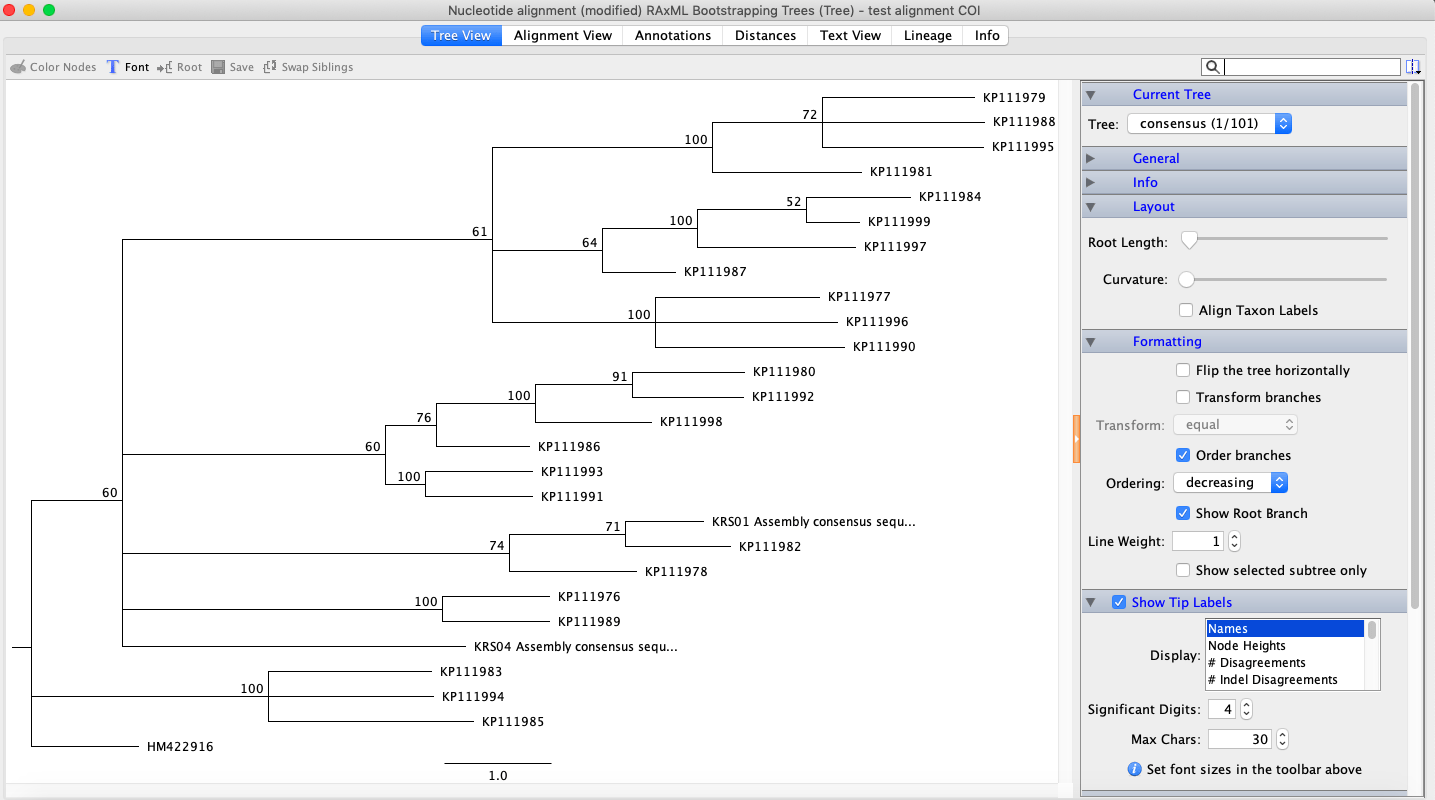
RAxML bootstrapping tree.
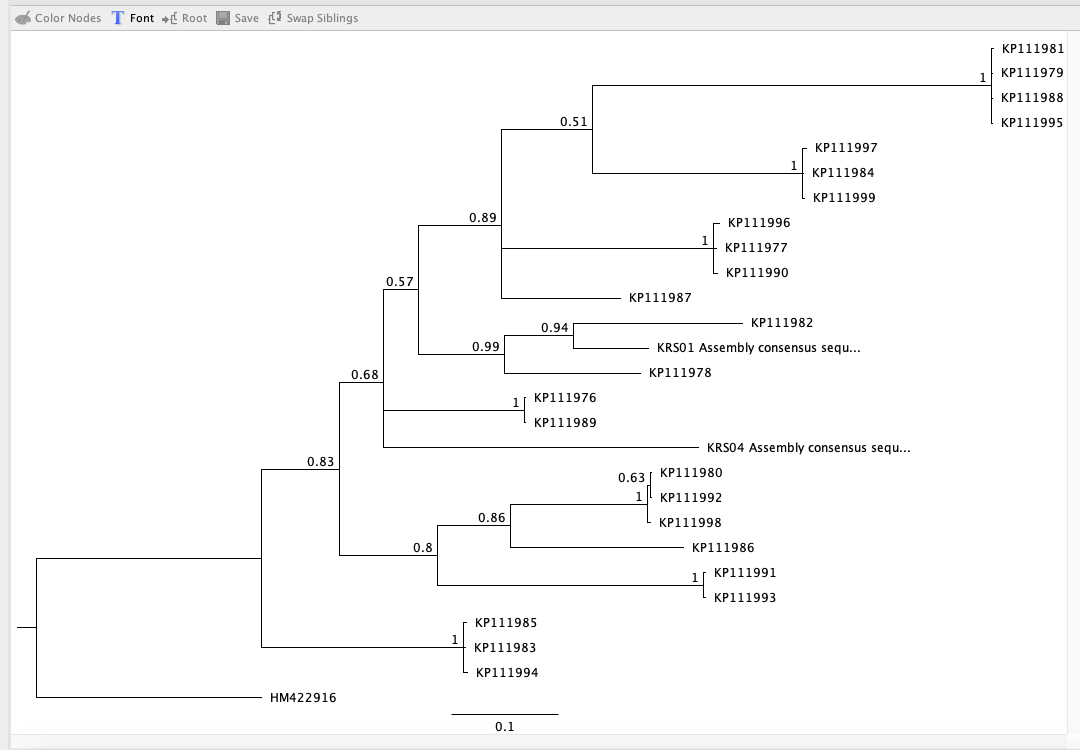
Initial Bayesian tree with bootstrap proportions.
