This lab is a continuation of Lab 02 and uses samples from that entry.
Materials:
- PCR tubes
- Pipette tips
- Distilled water
- Sap 10x
- SAP
- Exo
- Loading dye
- Ice
- Parafilm
- Gel electrophoresis equipment
Methods:
Part I – Gel Electrophoresis
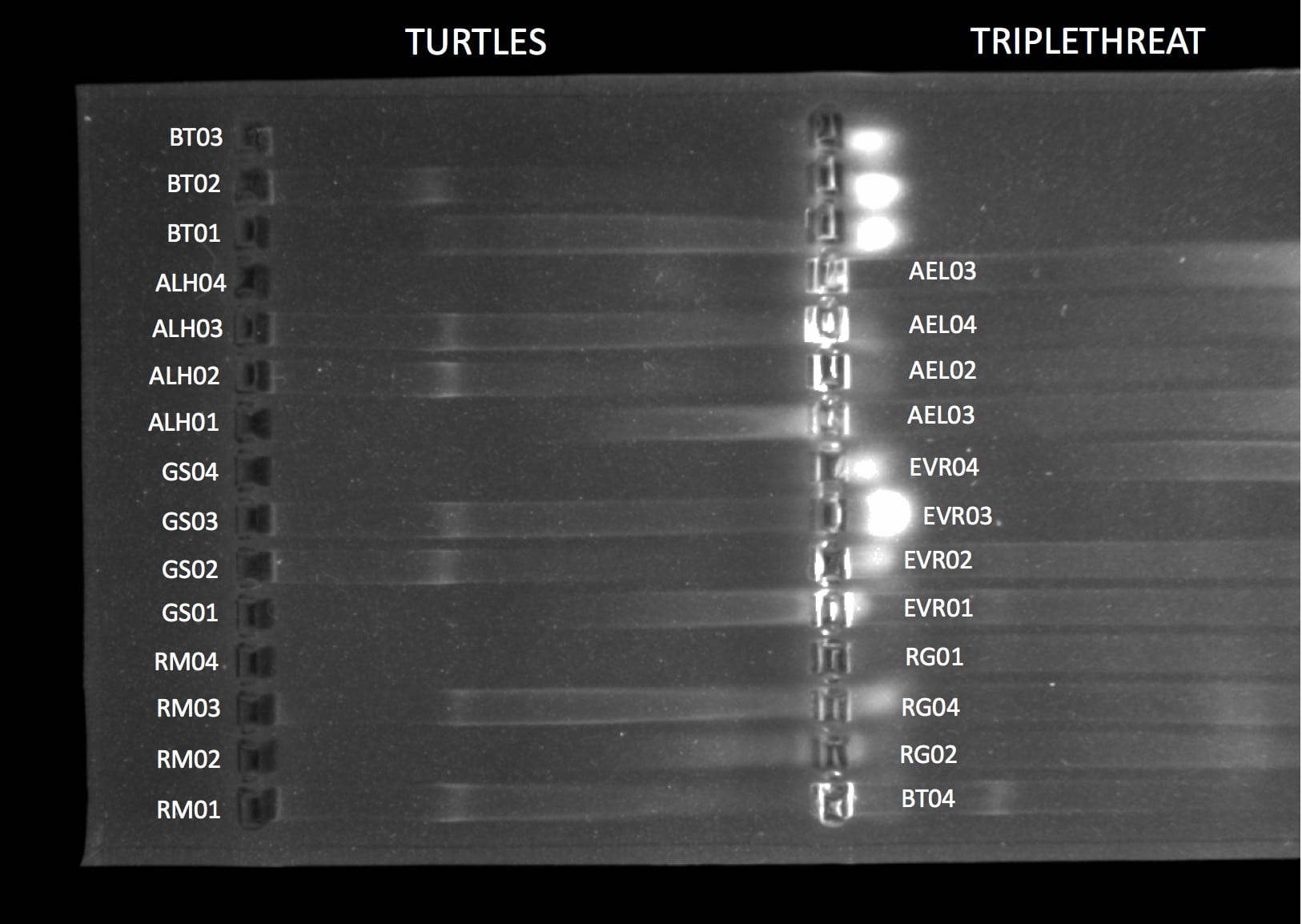
PCR tubes containing the samples labelled with EVR01, EVR02, EVR03, and EVR04 were thawed. 16 loading dye dots of about 1 uL were pipetted onto a sheet of parafilm. 3 uL of each sample was placed into its own dot. All dots were loaded into the gel alongside others’ samples as shown in Table 1. The gel was run at 130 volts for 30 minutes, which can be seen in Figure 1.
Part II – PCR Cleanup
The number of PCR cleanups that would need to be done for the products in Table 1 was calculated. Reagents were placed on ice. 7.5 uL of EVR01, EVR02, EVR03, and EVR04 were pipetted into their own 0.2 uL PCR tubes which were labeled with their respective codes. The ExoSap master mix was made using the numbers calculated in Table 2. 12.5 uL of the ExoSap mix was added to each PCR product tube. The tubes were placed into a thermocycler and the EXOSAP program was started which ran for about 45 minutes. The PCR tubes were placed into a labeled tube rack and placed in the freezer.
Table 1
Gel Electrophoresis Template: Genomic DNA PCR product
| Lane # (L -> R) | Sample ID # |
| TOP | |
| 1 | Negative control |
| 2 | RG01 |
| 3 | RG02 |
| 4 | RG04 |
| 5 | EVR01 |
| 6 | EVR02 |
| 7 | EVR03 |
| 8 | EVR04 |
| 9 | AEL01 |
| 10 | AEL02 |
| 11 | AEL03 |
| 12 | AEL04 |
| 13 | |
| 14 | |
| 15 | Ladder |
Table 2
Recipe to clean-up one PCR reaction of 7.5 uL
Master Mix: Rxn: 1 Rxns: 13
H2O 10.59 uL 137.67 (138)
10x buffer (Sap 10x) 1.25 uL 16.25 (16.3)
SAP 0.44 uL 5.72 (5.7)
Exo 0.22 uL 2.86 (2.9)
Master Mix Total 12.5 uL 162.5
PCR Product:
PCR 7.5 ul
Total Cleaned-up Volume 20.0 uL
Figure 1