Today we edited and aligned Mimulus cardinalis sequences in Geneious. Since our PCR attempts did not work, we worked with an older dataset. I worked with sequences from the 5536 EPIC marker.
1. First alignment
I did a muscle alignment of the sequences to do a first round of editing. First, I trimmed the ends to remove areas of poor quality. Next, I looked for polymorphisms and heterozygotes. There were about 10 polymorphic columns, and 3 heterozygote columns. Some sequences were not read as heterozygotes despite having two peaks, so I looked through the columns to edit any obvious heterozygotes with the correct code. Lastly, I edited any areas of obviously poor quality within the sequence. I then saved the alignment and edits to the sequences
2. Assemble forward and reverse reads and generate consensus sequences
I assembled each pair of forward and reverse reads. I then checked the assembly again for any area of poor quality or heterozygotes that I missed on the last step. I did not need to edit much since I edited on the previous step. Almost all of each assembly was of very high quality. I then generated the consensus sequences for all of the pairs. I then edited the names so that they were just the sample ID, so that they can be aligned with the same samples for the other EPIC markers. Two samples, JP1167 and the outlier M. lewisii sample, only had one read so I did not generate consensus sequences, but checked them for any necessary edits and used them in the alignment in the next step.
3. Final alignment
Using the consensus sequences, I built a MUSCLE alignment again. I looked at the alignment to see if any edits were again necessary, but I did not make any.
4. Infer Bayesian tree
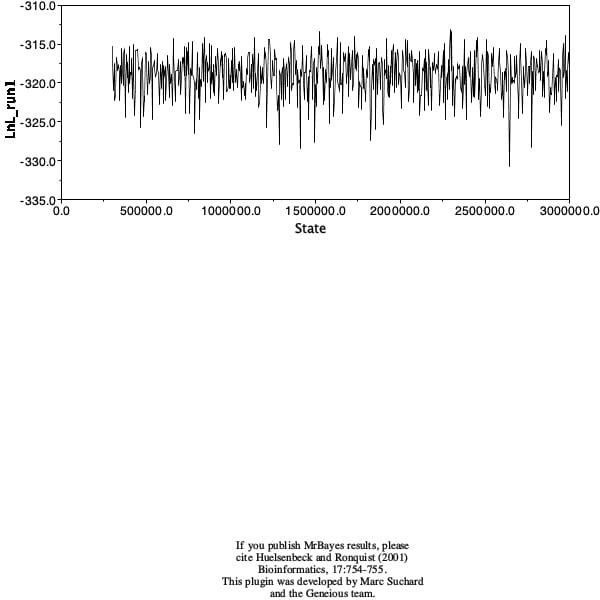
At home, I ran a bayesian analysis to infer a tree. I used TG0248 (M. lewisii) as the outgroup, and ran it with a chain length of 3,000,000, a burn-in length of 300,000, and a subsampling frequency of 500. My tree had no strongly supported clades and was one giant polytomy.