November 14, 2018
The wet lab consisted of two parts: the ISSR Analysis of the Lupinus aboreus , and the concatenation of Mimulus cardinalis alignments from the four EPIC markers.
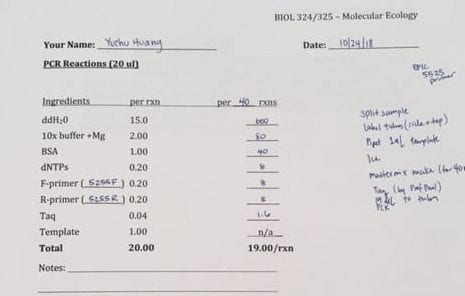
Part one of the lab, the ISSR analysis of the Lupinus aboreus samples, consisted of running the PCR reactions generated from previous labs. First, approximately 1-µL of loading dye was added to each of the PCR tubes, making sure to switch pipette tips between tubes. After completion, 5-µL of each sample was loaded onto the large 2% agarose gel by a tablemate. 48 samples from our table was loaded onto one of the rows of 50 wells, with the remaining 12 samples occupying the wells in the last row, following the group before us. The first and last wells of each row were meant to be saved for the ladder. Having established the order of our samples, it was determined that my samples would be loaded last. Thus, 12 of my samples should be on the last row with 3 on the row with my tablemates. The gels were then run on a low voltage of 20 volts. Due to circumstances, we did not get to score the banding patterns of the gel, though the protocol for identifying the bands would be to mark a ‘1’ if a band was present, and a ‘0’ if a band was absent.
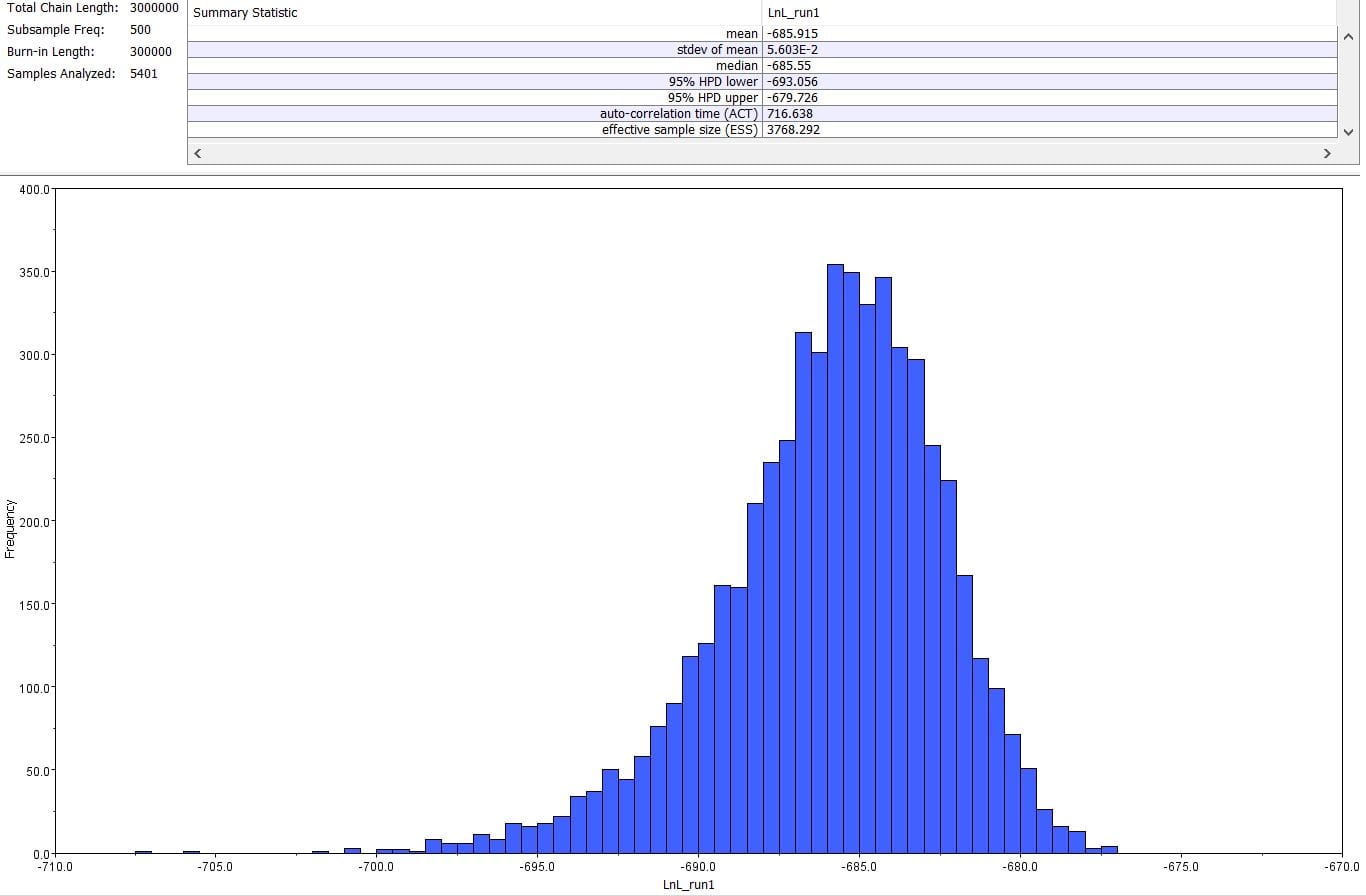
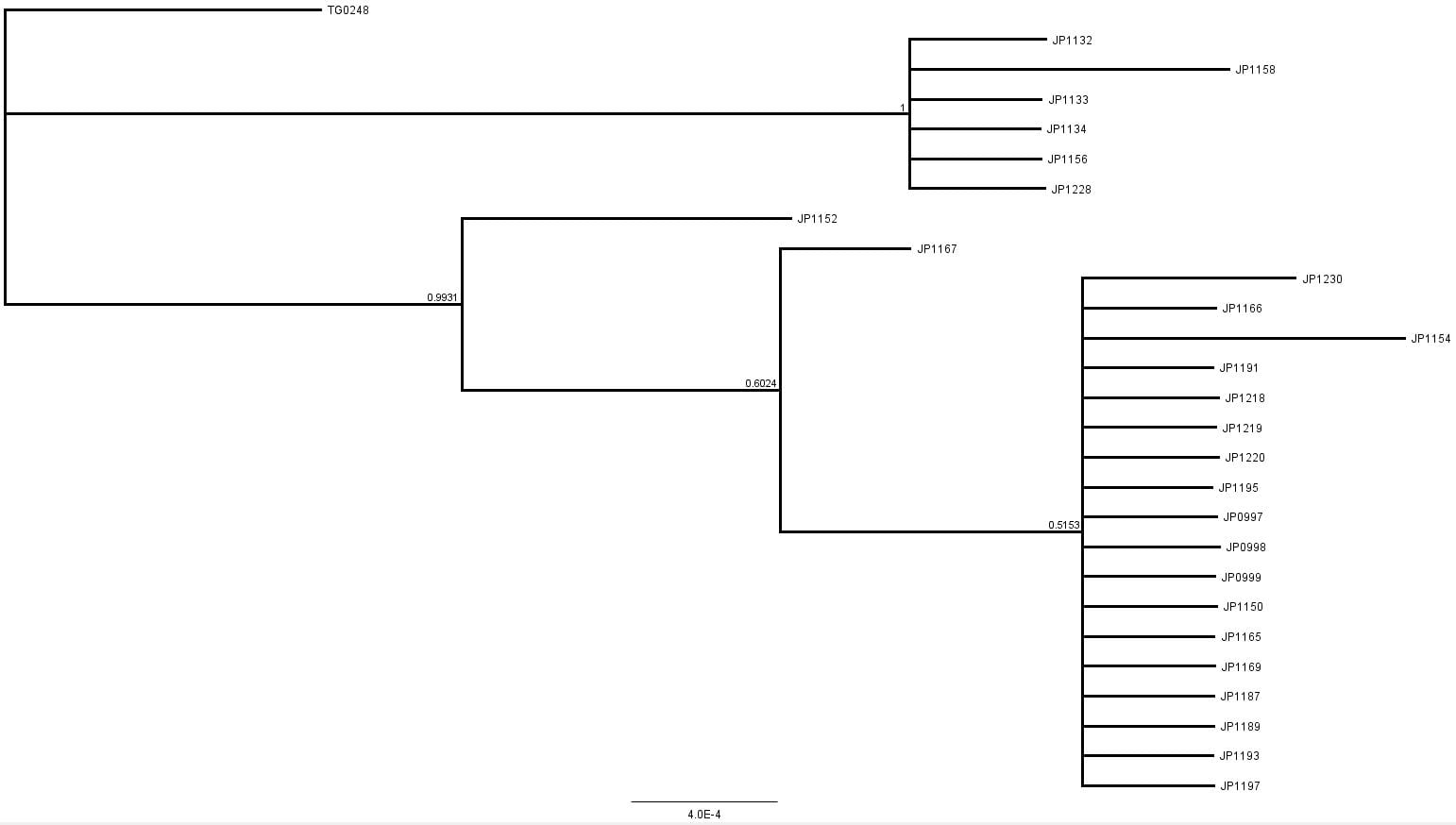
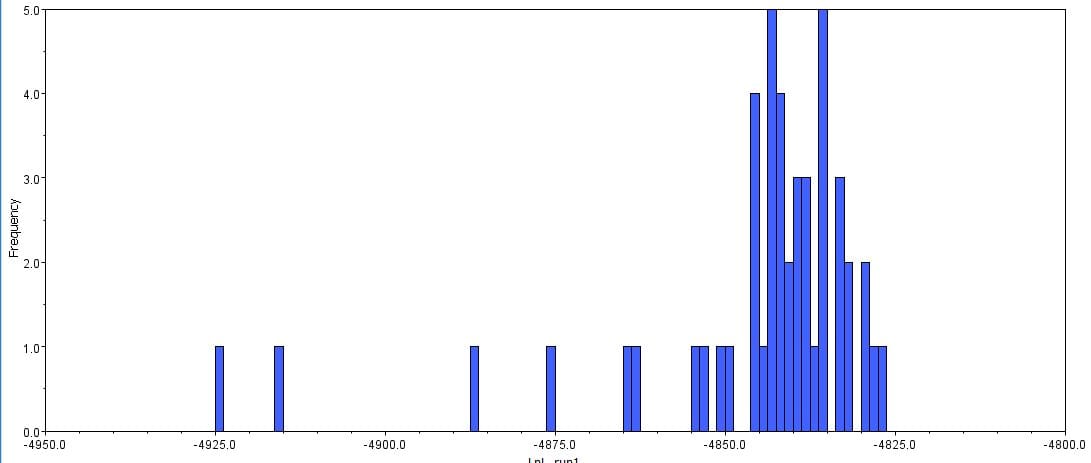
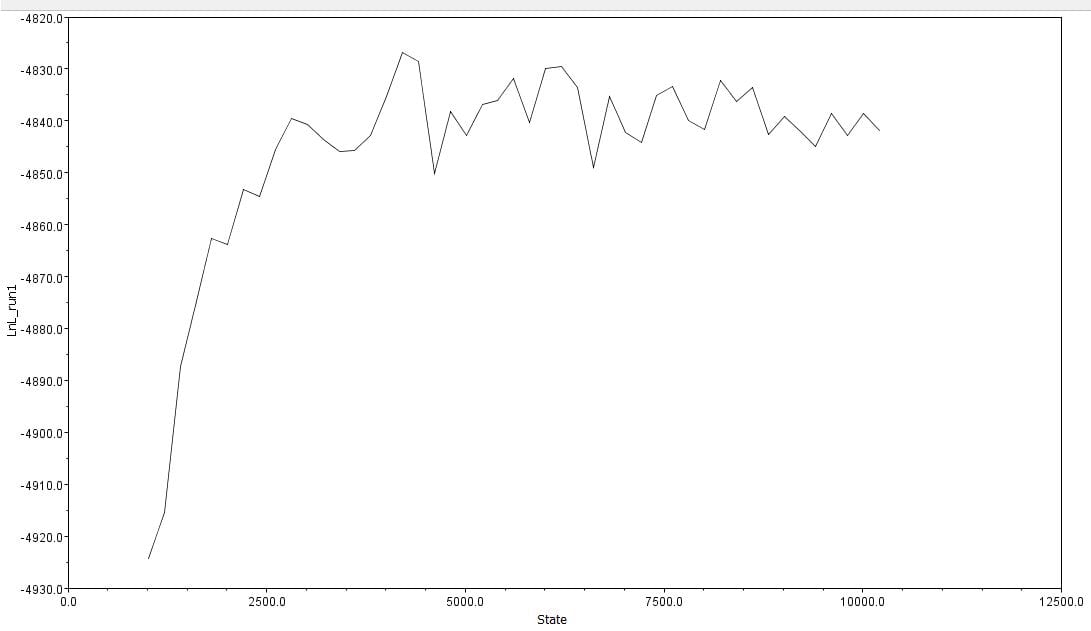
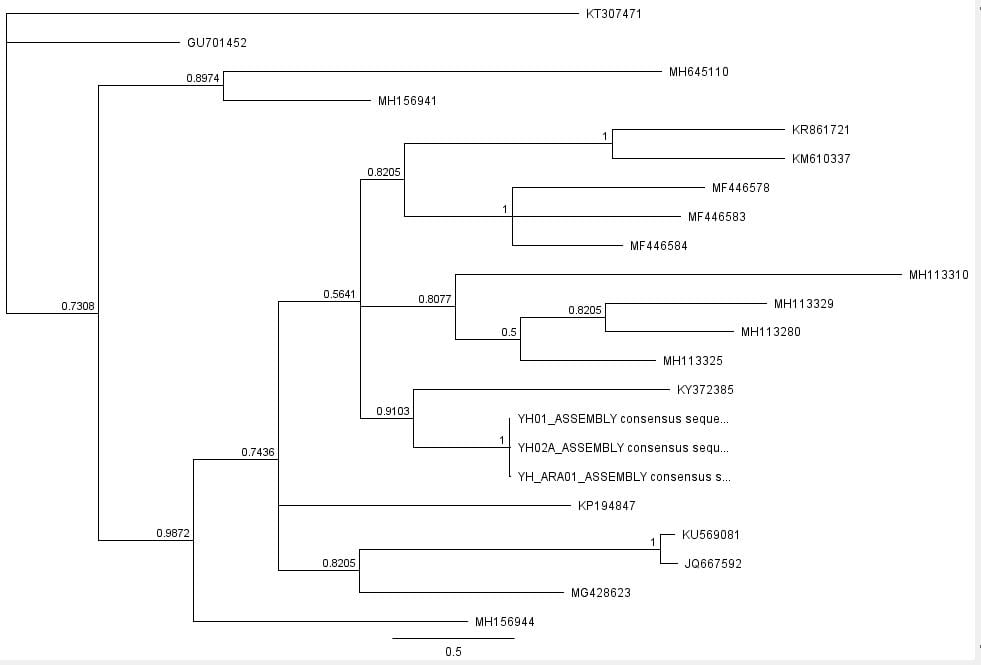
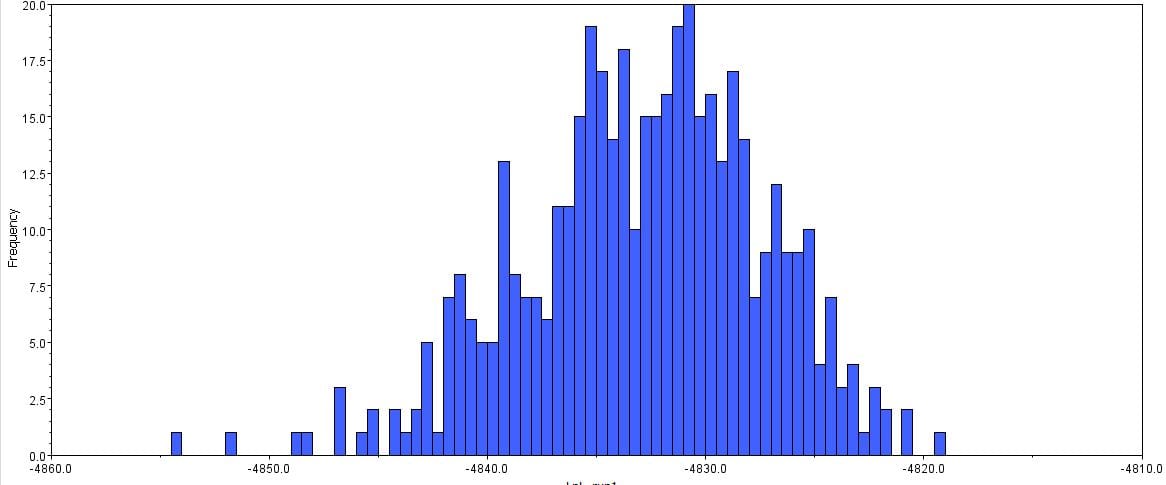
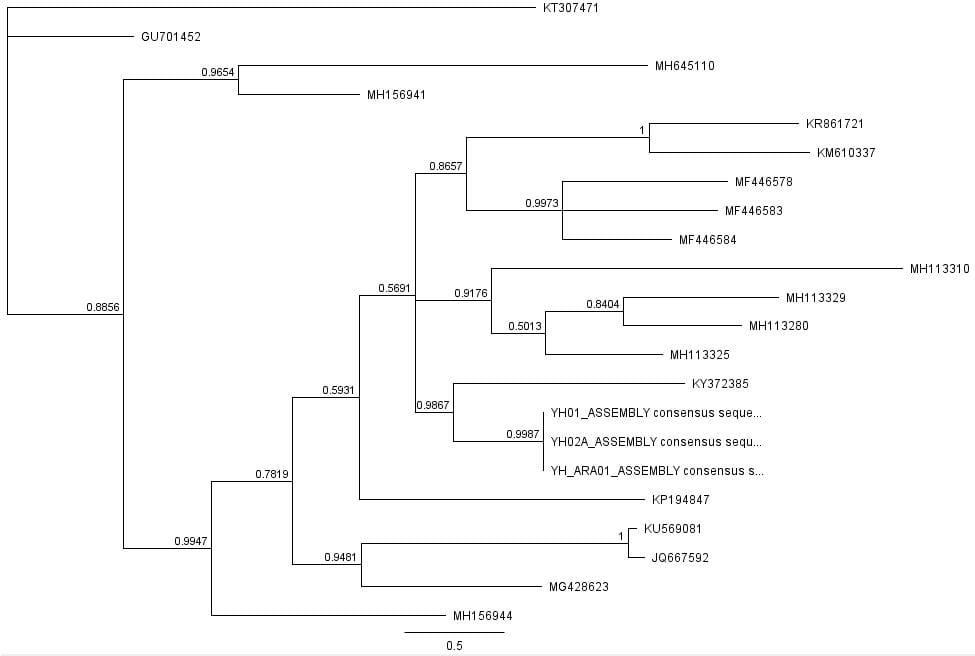
The second part of the lab was performed on Geneious. Having already identified heterozygous sites and built alignments from previous class data, several additional sequences were downloaded and imported into Geneious under the folder ‘5525 Consensus Sequence ver2’. These additional sequences were found on Canvas as the file ‘EPIC_5525_New’. Having imported the new marker documents, each forward and reverse pair was assembled together using the function ‘De Novo Assembly’. These consensus files were edited via trimming of bad starts and ends and relabeling of heterozygous or poorly called sites. Consensus sequences were generated from the edited forms of these assemblies. Using the ‘Batch Rename’ function under ‘Edit’ and selecting all the newly formed consensus sequences, 33 characters were erased to rename the files. The files were then copied and pasted into the ‘5525 Consensus Sequence ver2’ folder containing the previous alignment. All the consensus sequences were then selected (consisting of old and new) and a new Muscle nucleotide alignment was generated using default settings. Additional edits such as trimmings of bad ends and starts were performed before the file was saved and closed. The new alignment was then renamed to be ‘5525_Nucleotide alignment_YUCHUH’ containing a total of 33 sequences. A new folder was created under ‘Local’ called ‘Concatenate’. The new alignment was copied and pasted into this new folder. It was also exported as a geneious file and uploaded to Canvas under the assignment ‘EPIC ALIGNMENTS’. From the variety of marker alignments under the module ‘EPIC ALIGNMENTS’, three marker alignments generated by peers were imported into the ‘Concatenate’ folder. All four files in the ‘Concatenate’ folder was selected and the function ‘Concatenate Sequences or Alignments’ under ‘Tools’ was used. A file containing all four files was created called ‘Concatenated 1’. This file was then used for exercises later detailed.